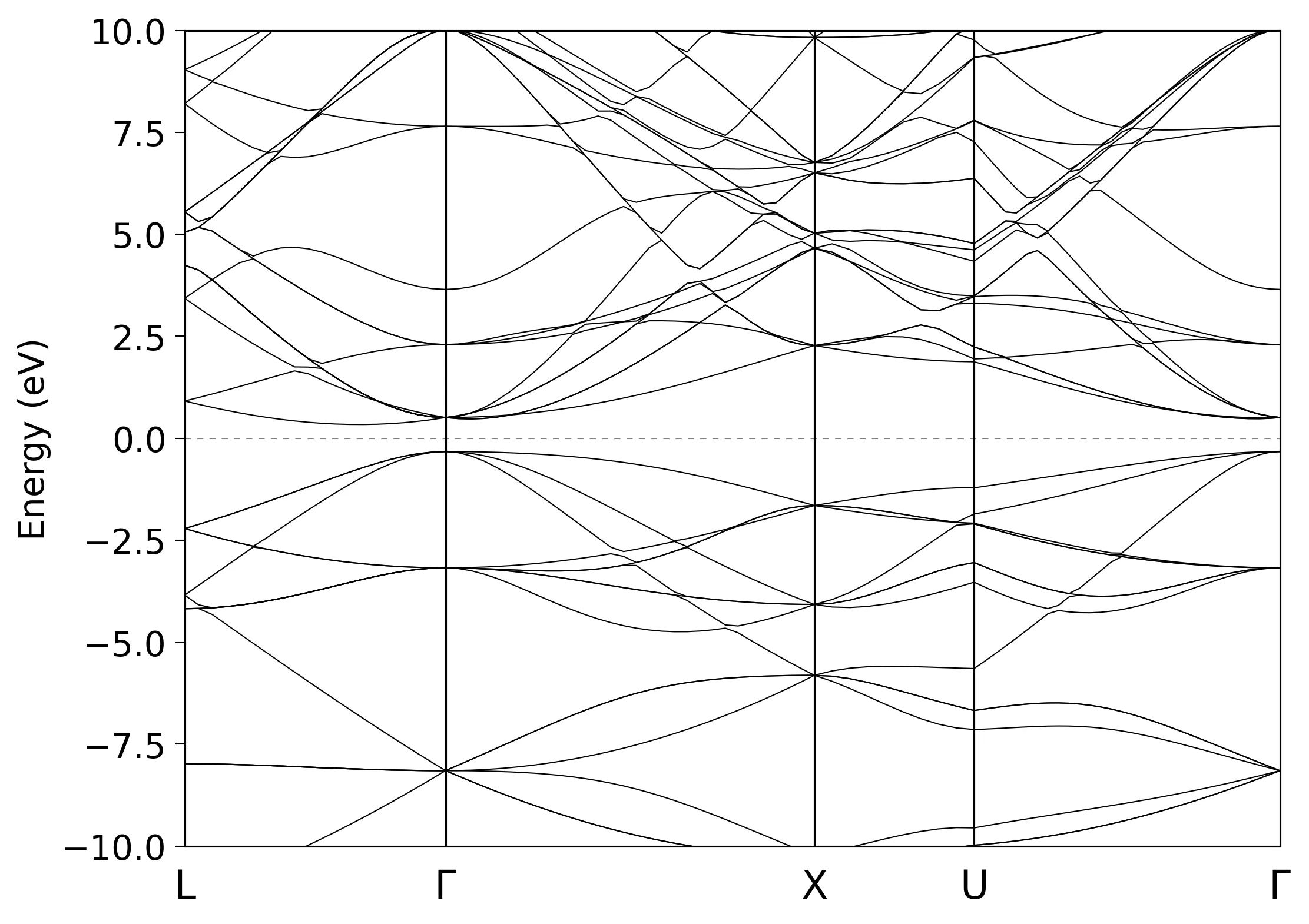
Bandstructure calculation for silicon
Here we will calculate the bandstructure of silicon. First step is to perform the scf calculation. Here is our input file:
src/silicon/silicon.in
# File Name
System.CurrrentDirectory ./ # default=./
System.Name silicon
DATA.PATH /home/svu/slspkd/openmx3.9/DFT_DATA19
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 0 # default=1 (0-2)
# Definition of Atomic Species
Species.Number 1
<Definition.of.Atomic.Species
Si Si7.0-s2p2d1 Si_PBE19
Definition.of.Atomic.Species>
# Atoms
Atoms.Number 8
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 Si 0.000000000000 0.000000000000 0.000000000000 2.0 2.0
2 Si 0.000000000000 2.715000000000 2.715000000000 2.0 2.0
3 Si 2.715000000000 0.000000000000 2.715000000000 2.0 2.0
4 Si 2.715000000000 2.715000000000 0.000000000000 2.0 2.0
5 Si 1.357500000000 1.357500000000 1.357500000000 2.0 2.0
6 Si 1.357500000000 4.072500000000 4.072500000000 2.0 2.0
7 Si 4.072500000000 1.357500000000 4.072500000000 2.0 2.0
8 Si 4.072500000000 4.072500000000 1.357500000000 2.0 2.0
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
5.430000000000 0.000000000000 0.000000000000
0.000000000000 5.430000000000 0.000000000000
0.000000000000 0.000000000000 5.430000000000
Atoms.UnitVectors>
# SCF or Electronic System
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW
scf.SpinPolarization off # On|Off
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 160.0 # default=150 (Ry)
scf.maxIter 100 # default=40
scf.EigenvalueSolver band # Recursion|Cluster|Band
scf.lapack.dste dstevx # dstegr|dstedc|dstevx, default=dstegr
scf.Kgrid 7 7 7 # means nk1xnk2xnk3
scf.Mixing.Type rmm-diisk # Simple|Rmm-Diis|Gr-Pulay
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.200 # default=0.40
scf.Mixing.History 15 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-8 # default=1.0e-6 (Hartree)
Band.dispersion on # on|off, default=off
Band.Nkpath 4
<Band.kpath
20 0.0000 0.5000 0.0000 0.0000 0.0000 0.0000 L G
30 0.0000 0.0000 0.0000 -0.500 0.0000 -0.500 G X
10 -0.500 0.0000 -0.500 -0.375 0.2500 -0.375 X U
30 -0.375 0.2500 -0.375 0.0000 0.0000 0.0000 U G
Band.kpath>
We can run the calculation using mpirun or submit via job-script:
mpirun -np 12 openmx silicon.in > silicon.out
While the calculation is running, you can monitor the convergence by:
grep "dUele" silicon.out | tail
Next step is to extract bandstructure data from silicon.Band file:
bandgnu13 silicon.Band
Finally, we can plot the bandstructure using gnuplot or any program of your choice:
gnuplot silicon.GNUBAND