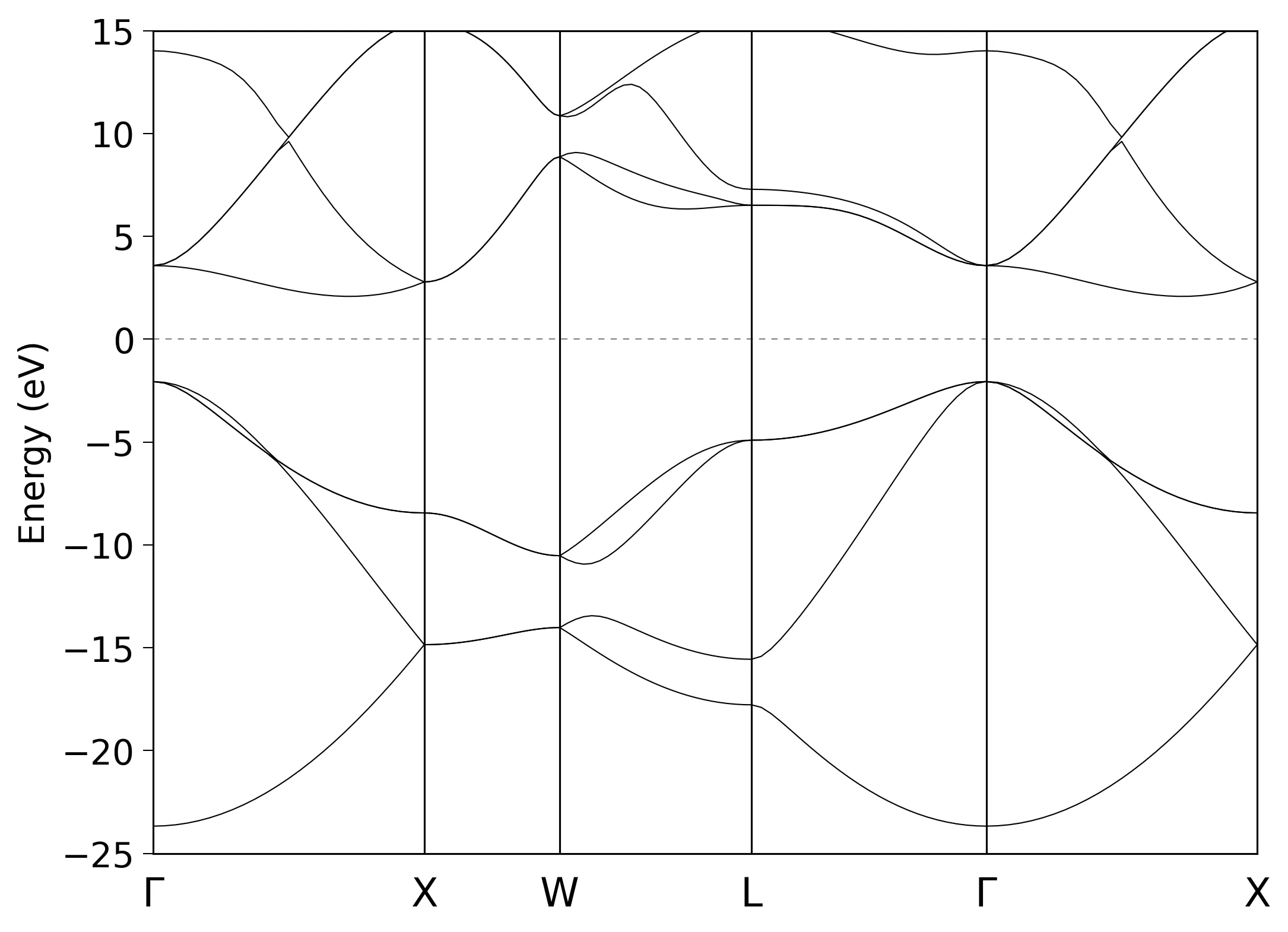
Bandstructure and DOS of diamond
We will run the bandstructure calculation from the OpenMX example file. Here is the input file:
src/diamond/diamond.in
# File Name
System.CurrrentDirectory ./ # default=./
System.Name diamond
DATA.PATH /home/svu/slspkd/openmx3.9/DFT_DATA19
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 0 # default=1 (0-2)
# Definition of Atomic Species
Species.Number 1
<Definition.of.Atomic.Species
C C5.0-s2p2d1 C_CA19
Definition.of.Atomic.Species>
# Atoms
Atoms.Number 2
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates
1 C 0.000 0.000 0.000 2.0 2.0
2 C 0.890 0.890 0.890 2.0 2.0
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors
1.7800 1.7800 0.0000
1.7800 0.0000 1.7800
0.0000 1.7800 1.7800
Atoms.UnitVectors>
# SCF or Electronic System
scf.XcType LDA # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 150.0 # default=150 (Ry)
scf.maxIter 100 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 7 7 7 # means n1 x n2 x n3
scf.Mixing.Type rmm-diisk # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.30 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.700 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-10 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 1 # default=0.5 (fs)
MD.Opt.criterion 1.0e-5 # default=1.0e-4 (Hartree/bohr)
# Band dispersion
#
Band.dispersion on # on|off, default=off
<Band.KPath.UnitCell
3.56 0.00 0.00
0.00 3.56 0.00
0.00 0.00 3.56
Band.KPath.UnitCell>
# if <Band.KPath.UnitCell does not exist,
# the reciprical lattice vector is employed.
Band.Nkpath 5
<Band.kpath
25 0.0 0.0 0.0 1.0 0.0 0.0 g X
25 1.0 0.0 0.0 1.0 0.5 0.0 X W
25 1.0 0.5 0.0 0.5 0.5 0.5 W L
25 0.5 0.5 0.5 0.0 0.0 0.0 L g
25 0.0 0.0 0.0 1.0 0.0 0.0 g X
Band.kpath>
# MO output
#
MO.fileout off # on|off
num.HOMOs 1 # default=2
num.LUMOs 1 # default=2
MO.Nkpoint 2 # default=1
<MO.kpoint
0.0 0.0 0.0
0.0 0.0 0.2
MO.kpoint>
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -25.0 20.0 # default = -20 20
Dos.Kgrid 12 12 12 # default = Kgrid1 Kgrid2 Kgrid3
FermiSurfer.fileout on
We can run the calculation using mpirun or submit via job-script:
mpirun -np 12 openmx diamond.in > diamond.out
Next step is to extract bandstructure data from diamond.Band file:
bandgnu13 diamond.Band
Finally, we can plot the bandstructure using gnuplot or any plotting program of your choice:
gnuplot diamond.GNUBAND
Density of States (DOS)
DosMain diamond.Dos.val diamond.Dos.vec
It will ask few options interactively. Use of tetrahedron method is suitable.
The output file diamond.DOS.Tetrahedron will contain the energy, DOS and
integrated DOS columns.
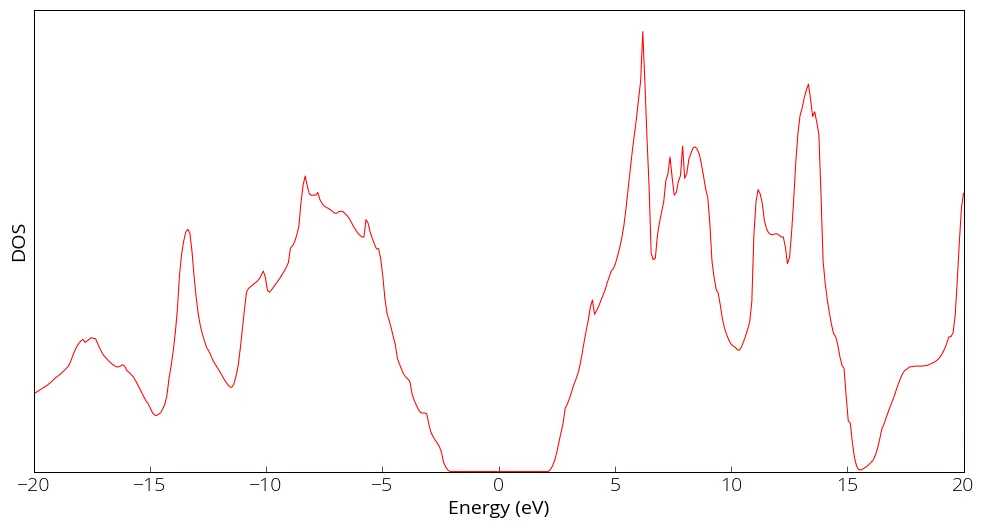
While running the DosMain program, you can also choose PDOS for projected density of states for each orbitals.