DFT+U calculation
We will calculate the density of states for NiO with and without including Hubbard . We will start from the example input file for NiO (check work directory under OpenMX installation).
src/NiO/NiO.in
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name nio
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (1-3)
DATA.PATH /home/svu/slspkd/openmx3.9/DFT_DATA19
# Definition of Atomic Species
#
Species.Number 2
<Definition.of.Atomic.Species
Ni Ni6.0S-s2p2d2f1 Ni_CA19S
O O5.0-s2p2d1 O_CA19
Definition.of.Atomic.Species>
<Hubbard.U.values # eV
Ni 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 5.0 2d 0.0 1f 0.0
O 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 0.0
Hubbard.U.values>
<Hund.J.values # eV
Ni 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 0.5 2d 0.0 1f 0.0
O 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 0.0
Hund.J.values>
# Atoms
#
Atoms.Number 4
Atoms.SpeciesAndCoordinates.Unit AU # Ang|AU
<Atoms.SpeciesAndCoordinates
1 Ni 0.0 0.0 0.0 9.5 6.5 off
2 Ni 3.94955 3.94955 0.0 6.5 9.5 off
3 O 3.94955 0.0 0.0 3.0 3.0 off
4 O 3.94955 3.94955 3.94955 3.0 3.0 off
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit AU # Ang|AU
<Atoms.UnitVectors
7.89910 3.94955 3.94955
3.94955 7.89910 3.94955
3.94955 3.94955 7.89910
Atoms.UnitVectors>
# SCF or Electronic System
#
scf.XcType LSDA-CA # LDA|LSDA-CA|GGA-PBE
# DFT+U part #
scf.Hubbard.U On # On|Off, default=off
scf.Hubbard.Occupation dual # onsite|full|dual, default=dual
scf.DFTU.Type 2 # 1:Simplified(Dudarev)|2:General, default=1
scf.dc.Type sFLL # sFLL|sAMF|cFLL|cAMF, default=sFLL
scf.Slater.Ratio 0.625 # default=0.625
scf.Yukawa off # default=off
scf.SpinPolarization On # On|Off
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 150.0 # default=150 (Ry)
scf.maxIter 200 # default=40
scf.EigenvalueSolver band # Recursion|Cluster|Band
scf.Kgrid 6 6 6 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay
scf.Init.Mixing.Weight 0.20 # default=0.30
scf.Min.Mixing.Weight 0.01 # default=0.001
scf.Max.Mixing.Weight 0.30 # default=0.40
scf.Kerker.factor 1.00 # default=1.00
scf.Mixing.History 5 # default=5
scf.Mixing.StartPulay 6 # default=6
scf.criterion 1.0e-7 # default=1.0e-6 (Hartree)
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|DIIS|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.05 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
# Band dispersion
#
Voronoi.Charge on
Band.dispersion off # on|off, default=off
# if <Band.KPath.UnitCell does not exist,
# the reciprical lattice vector is employed.
Band.Nkpath 1
<Band.kpath
25 0.0 0.0 0.5 0.0 0.5 0.5 Z K
Band.kpath>
# MO output
#
MO.fileout off # on|off
num.HOMOs 1 # default=1
num.LUMOs 1 # default=1
MO.Nkpoint 1 # default=1
<MO.kpoint
0.0 0.0 0.0
MO.kpoint>
# DOS and LDOS
#
Dos.fileout on # on|off , default=off
Dos.Erange -10.0 10.0 # default = -20 20
Dos.Kgrid 9 9 9 # default = Kgrid1 Kgrid2 Kgrid3
# output Hamiltonian and overlap
#
HS.fileout off # on|off, default=off
note
If the initial spin configuration is unpolarized for LDA+U calculations, it is
required to provide the switch for enhancement of orbital polarization in the
LDA+U method in the last column of Atoms.SpeciesAndCoordinates, on means
that the enhancement is made, off means no enhancement.
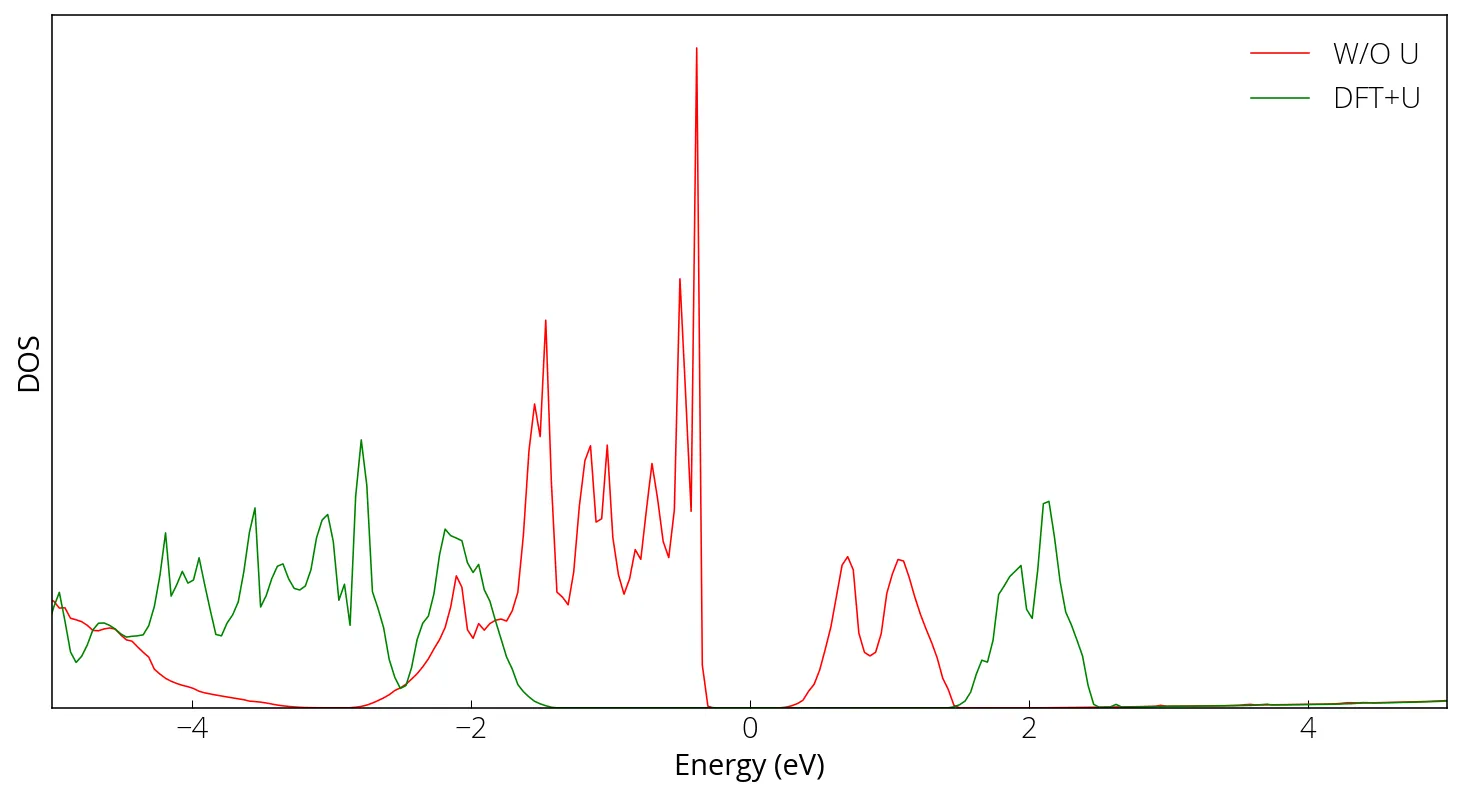
Here we compare the density of states with and without considering electronic correlation.
tip
If you have difficulty with SCF convergence, you might try
scf.Mixing.Type Rmm-Diish
which is suitable for the plus U method and the constraint schemes. Find more details here.