Spin-Orbit Coupling
In order to consider spin orbit coupling effect in our electronic structure
calculation in quantum espresso, we need to use a full relativistic pseudo
potential. Following settings are needed in the &SYSTEM card:
&SYSTEM
...
noncolin = .true.
lspinorb = .true.
...
/
Non collinear spin
In simple spin polarized calculation (nspin=2), the spin quantum number (up or
down) is considered in the calculation. In non-collinear case, the spin has more
degrees of freedom, and can be oriented in any direction. Non-collinear
magnetism is quite common in nature, where the spins are not parallel
(ferromagnetic) or anti-parallel (antiferromagnetic), rather they orient in
spirals, helicoids, canted or disordered. Non-collinear magnetism can occur
because of geometric frustration of magnetic interaction. It can also occur due
to the magnetocrystalline anisotropy which is the result of interaction between
the spin and lattice interaction. This relativistic effect comes via spin-orbit
coupling.
We can constrain the magnetic moment:
&SYSTEM
...
constrained_magnetization = 'atomic direction'
...
/
Starting magnetization can be specified by angle1 (angle with axis) and
angle2 (angle of projection in -plane and with -axis). Also check the
penalty function ().
&SYSTEM
...
angle1(i) = 0.0d0
angle2(i) = 0.0d0
lambda = 0.5
...
/
i is the index of the atom in ATOMIC_SPECIES card.
Strategy for convergence
Spin-orbit coupling calculations are often hard to converge. Use a smaller
mixing_beta for such calculations. First perform a collinear calculation with
non-relativistic pseudopotential, and then start from the obtained charge
density to perform non-colinear spin orbit calculation.
&ELECTRONS
...
mixing_beta = 1.0000000000d-01
startingpot = 'file'
/
When starting with non-collinear calculation from an existing charge density
file from a collinear lsda calculation, we need to set lforcet=.true.. It
assumes previous density points in z direction, and rotates in the direction
specified by angle1 (initial magnetization angle with -axis in degrees),
and angle2 (angle in degrees for projections in -plane and with -axis).
&SYSTEM
...
angle1(i) = 0.0
angle2(i) = 0.0
lforcet = .true.
/
Also, make sure that energy and charge density cutoffs are sufficient. Certain pseudo potentials might have issues, try with pseudo potentials from a different library. In case of metallic systems, remember to apply smearing.
-
S matrix not positive definite: This error might appear due to numerical instability from overlapping atoms. Check atomic positions carefully. In one my calculations, this error was resolved after setting higher
ecutrho. -
Simplified LDA+U not implemented with
noncolmagnetism, uselda_plus_u_kind=1.
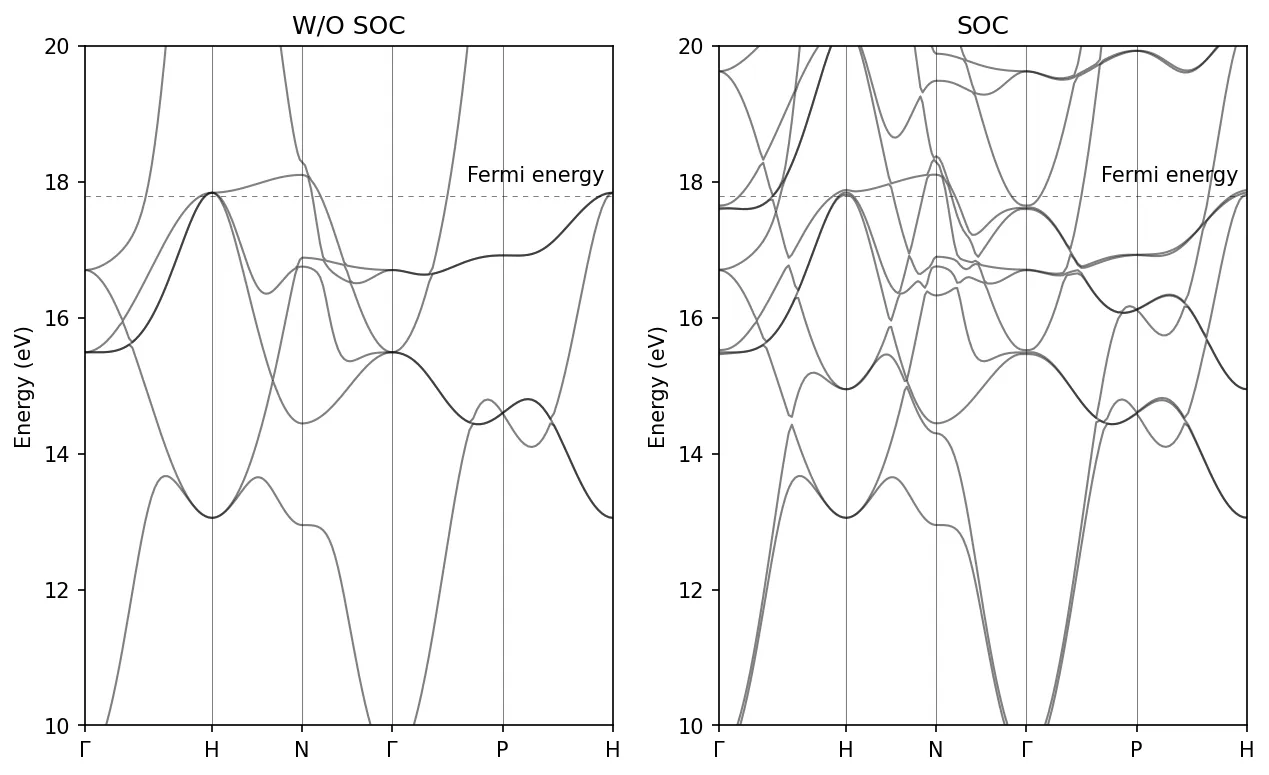
Bandstructure of Fe with SOC
&control
calculation='scf'
pseudo_dir = '../pseudos/',
outdir='./tmp/'
prefix='fe'
/
&system
ibrav = 3,
celldm(1) = 5.39,
nat= 1,
ntyp= 1,
noncolin=.true.,
lspinorb=.true.,
starting_magnetization(1)=0.3,
ecutwfc = 70,
ecutrho = 850.0,
occupations='smearing',
smearing='marzari-vanderbilt',
degauss=0.02
/
&electrons
diagonalization='david'
conv_thr = 1.0e-8
mixing_beta = 0.7
/
ATOMIC_SPECIES
Fe 55.845 Fe.rel-pbe-spn-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS alat
Fe 0.0 0.0 0.0
K_POINTS AUTOMATIC
14 14 14 1 1 1
Run the scf calculation:
mpirun -np 8 pw.x -i pw.scf.fe_soc.in > pw.scf.fe_soc.out
Prepare the input file for nscf bands calculation:
&control
calculation='bands'
pseudo_dir = '../pseudos/',
outdir='./tmp/'
prefix='fe'
/
&system
ibrav = 3,
celldm(1) = 5.39,
nat= 1,
ntyp= 1,
noncolin=.true.,
lspinorb=.true.,
starting_magnetization(1)=0.3,
ecutwfc = 70,
ecutrho = 850.0,
occupations='smearing',
smearing='marzari-vanderbilt',
degauss=0.02
/
&electrons
diagonalization='david'
conv_thr = 1.0e-8
mixing_beta = 0.7
/
ATOMIC_SPECIES
Fe 55.845 Fe.rel-pbe-spn-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS alat
Fe 0.0 0.0 0.0
K_POINTS tpiba_b
6
0.000 0.000 0.000 40 !gamma
0.000 1.000 0.000 40 !H
0.500 0.500 0.000 30 !N
0.000 0.000 0.000 30 !gamma
0.500 0.500 0.500 30 !P
0.000 1.000 0.000 1 !H
Run the bands calculation:
mpirun -np 8 pw.x -i pw.bands.fe_soc.in > pw.bands.fe_soc.out
Finally post process the bandstructure data:
&BANDS
outdir='./tmp/',
prefix='fe',
filband='fe_bands_soc.dat',
/
In this case spin_component has been removed and we add lsigma(3)=.true.
that instructs the program to compute the expectation value for the z
component of the spin operator for each eigenfunction and save all values in
the file fe.noncolin.data.3. All values in this case are either +1/2 or -1/2.
mpirun -np 8 bands.x -i pp.bands.fe_soc.in > pp.bands.fe_soc.out
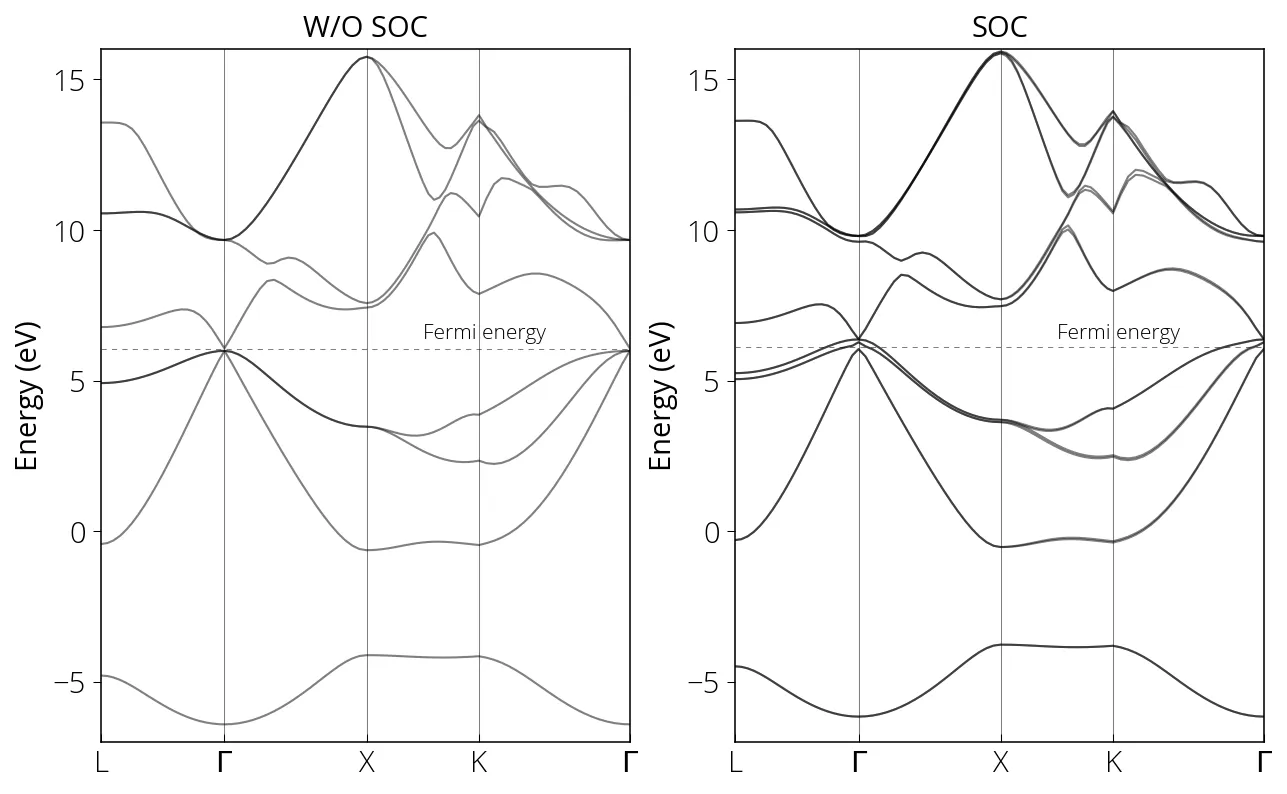
SOC calculation for GaAs
Please check the respective input files.
mpirun -np 8 pw.x -i pw.scf.GaAs_soc.in > pw.scf.GaAs_soc.out
mpirun -np 8 pw.x -i pw.bands.GaAs_soc.in > pw.bands.GaAs_soc.out
mpirun -np 8 bands.x -i pp.bands.GaAs_soc.in > pp.bands.GaAs_soc.out