Magnetic system: bulk iron
I am following this example from the ICTP online school 2021.
We will perform the SCF KS orbital calculations on magnetic (nspin=2) iron.
Since the d-orbitals of Fe atom are localized/
If we have crystal structure with only one atom per unit cell, or only one type of atoms, the only possible ordering is ferromagnetic. In such cases, we need to form supercell with more number of atoms or label multiple atoms separately, so that their magnetic orientation could be different thus having the possibility of ferro- or antiferromagnetic final states.
Run the SCF calculations for both ferro- and antiferromagnetic structures. Notice that for ferromagnetic, we have BCC structure with only one type of atom, while we use simple cubic structure for antiferromagnetic case with two different atomic labels. For antiferromagnetic calculation, we need to start with opposite initial spins.
pw.x -i pw.scf.fe_fm.in > pw.scf.fe_fm.out
pw.x -i pw.scf.fe_afm.in > pw.scf.fe_afm.out
In case of the AFM calculation, if we have started with FM (say, for both atom
types starting_magnetization=0.6 ), the calculation would still converge to
AFM state as it is the true ground state for this system, albeit it would take
more iteration to converge. If a system has complex potential surface with local
minima, it it possible to get different final state magnetization depending on
the starting magnetization. In such cases, a stricter convergence criteria might
help.
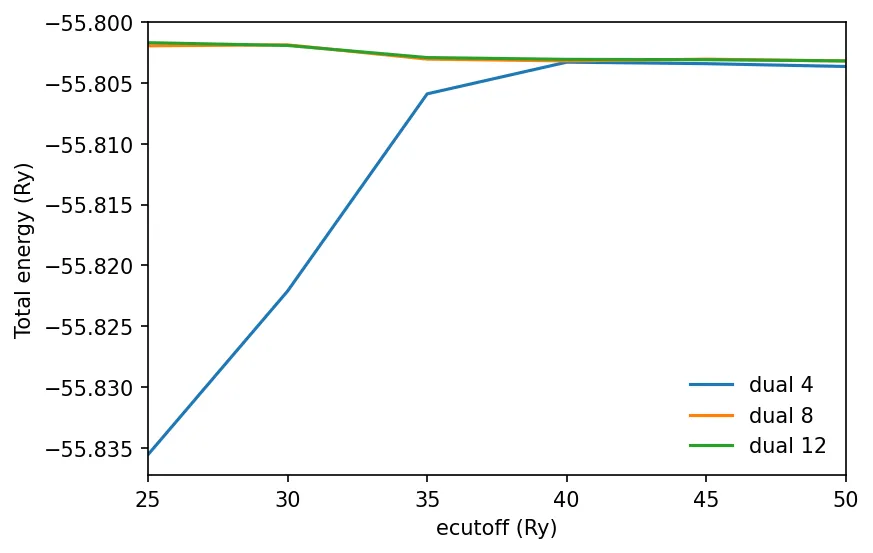
In case of ultrasoft pseudo potentials, the Quantum Espresso default of
ecutrho 4 times of ecutoff is not sufficient. We need to set ecutrho 8 or
even 12 times that of ecutoff. We must test the convergence for our set
values.
Convergence test for USPP
Below is the PWTK script file:
# load the pw.x input from file
load_fromPWI fe_scf_fm.in
# dual is the ratio ecutrho/ecutwfc
foreach dual { 4 8 12 } {
set fid [open etot-vs-ecutwfc.dual$dual.dat w]
foreach ecutwfc [seq 25 5 50] {
set name pw.Fe.scf.ecutwfc-$ecutwfc.dual-$dual
SYSTEM "ecutwfc = $ecutwfc
ecutrho = $ecutwfc*$dual "
runPW $name.in
set Etot [pwo_totene $name.out]
puts $fid "$ecutwfc $Etot"
}
close $fid
}
Run the script:
pwtk fe_ecut.pwtk
Density of states calculation
PWTK script to calculate DOS and p-DOS:
load_fromPWI fe_scf_fm.in
SYSTEM " ecutwfc = 40
ecutrho = 320 "
set name Fe
runPW pw.$name.scf.in
CONTROL { calculation = 'nscf' }
SYSTEM { occupations = 'tetrahedra' ,
degauss = , ! this is how variable is unset in PWTK
}
K_POINTS automatic {
12 12 12 1 1 1
}
runPW pw.$name.nscf.in
DOS "
fildos = '$name.dos.dat'
Emin = 5.0
Emax = 20.0,
DeltaE = 0.1
"
runDOS dos.$name.in
PROJWFC "
filpdos = '$name.pdos.dat'
Emin = 5.0
Emax = 20.0,
DeltaE = 0.1
"
runPROJWFC projwfc.$name.in
Below is the plots of total and projected density of states.
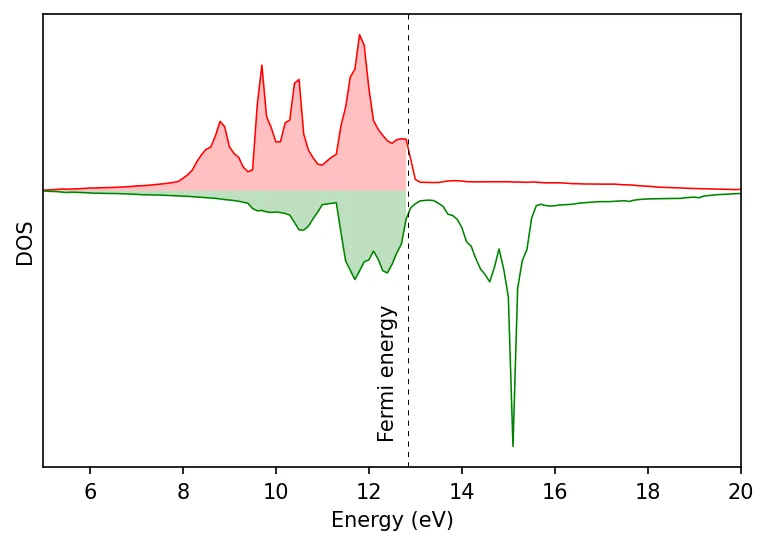
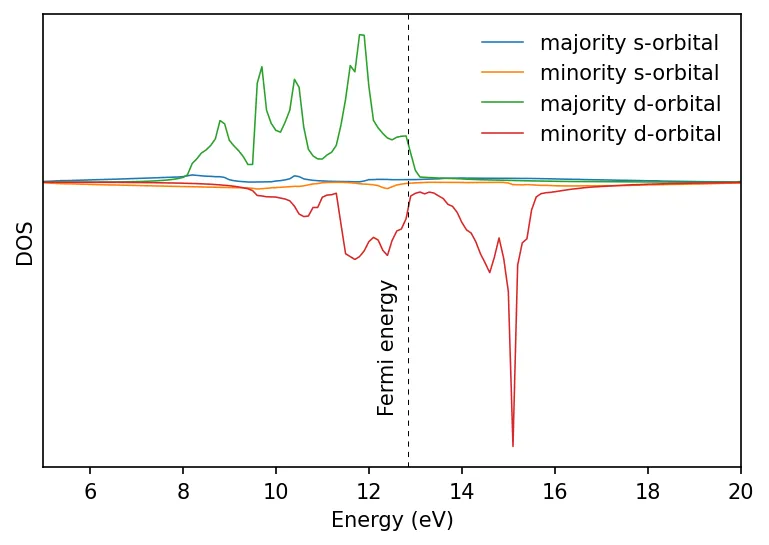
Also see bandstructure of Fe with and without SOC.
Paramagnetism
Paramagnetic materials have fluctuating magnetic moments that may not be properly described DFT. One approach is to model paramagnetic materials in DFT calculation by building a large supercell and assign randomly oriented magnetic moments.
Also note that DFT assumes zero temperature, so it makes sense to perform FM or AFM calculation for magnetic systems.
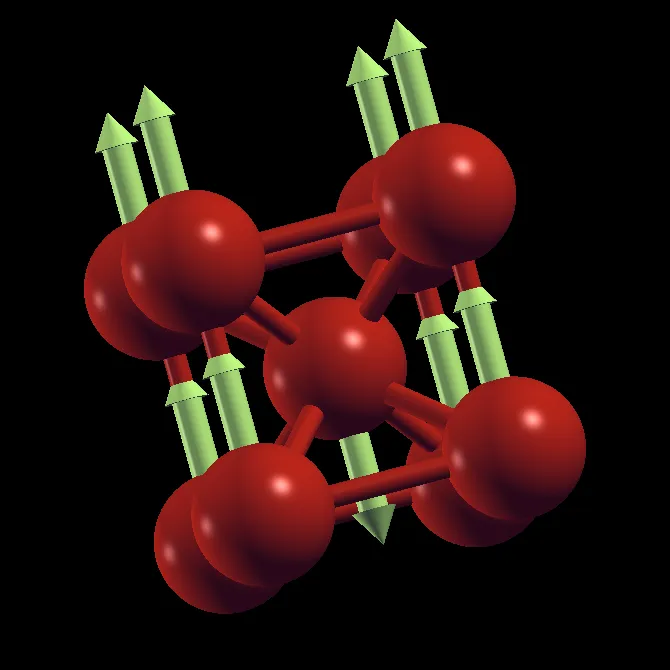
Visualizing magnetic moments
We can use XCrySDen to visualize the orientation of
magnetic moments. XCrySDen cannot directly read the Quantum Espresso output
files for magnetic moment vectors, instead we need to create the input .xsf
file with magnetic moments as force vector. You can also change the background
color from black from the Palette Menu which is located in the left of File
menu.
# this is a specification for crystal structure
CRYSTAL
# primitive lattice vectors (in Angstroms)
PRIMVEC
2.8681404710 0.0000000000 0.0000000000
0.0000000000 2.8681404710 0.0000000000
0.0000000000 0.0000000000 2.8681404710
# conventional lattice vectors (in Angstroms)
CONVVEC
2.8681404710 0.0000000000 0.0000000000
0.0000000000 2.8681404710 0.0000000000
0.0000000000 0.0000000000 2.8681404710
# First number stands for number of atoms in primitive cell
# the second number is always 1 for PRIMCOORD coordinates
# followed by atomic coordinates (in Angstroms) and forces:
# AtNum X Y Z Fx Fy Fz
PRIMCOORD
2 1
26 0.0000000000 0.0000000000 0.0000000000 0.00 0.00 0.01
26 1.4340702350 1.4340702350 1.4340702350 0.00 0.00 -0.01
Open the file from XCrySDen Menu: File → Open Structure → Open XSF. Then go to Display menu and select Forces. If you want to adjust scaling for the force vectors, go to Modify → Force Settings and set suitable Length factor.