Density of States calculation
Electronic density of states is an important property of a material.
= number of electronic states in the energy interval
Before we can run the Density of States (DOS) calculation, we need
-
Perform fixed-ion self consistent filed (scf) calculation. In plane-wave based DFT calculations the electronic density is expressed by functions of the form with energy given by .
-
Perform non-self consistent field (nscf) calculation with denser k-point grid. A large number of points are required DOS calculation, as the accuracy of DOS depends on the integration in space.
-
Finally, the DOS can be determined by integrating the electron density in space.
I have created a new input file (pw.scf.silicon_dos.in) which is very much the same as
our previous scf input file except some parameters are modified. You can find
all the input files in my GitHub repository. We used the lattice constant value that
we obtained from the relaxation calculation. We should not directly use the
experimental/real lattice constant values. Depending on the method and
pseudo-potential, it might result stress in the system. We have increased the
ecutwfc to have better precision. We run the scf calculation:
pw.x < pw.scf.silicon_dos.in > pw.scf.silicon_dos.out
Next, we have prepared the input file for the nscf calculation. Where is have
added occupations in the &system card as tetrahedra (appropriate for DOS
calculation). We have increased the number of k-points to 12 × 12 × 12 with
automatic option. Also specify nosym = .TRUE. to avoid generation of
additional k-points in low symmetry cases. outdir and prefix must be the
same as in the scf step, some of the inputs and output are read from previous
step. Here we can specify a larger number of nbnd to calculate unoccupied
bands. Number of occupied bands can be found in the scf output as number of
Kohn-Sham states.
pw.x < pw.nscf.silicon_dos.in > pw.nscf.silicon_dos.out
Now our final step is to calculate the density of states. The DOS input file as follows:
&DOS
prefix='silicon',
outdir='./tmp/',
fildos='si_dos.dat',
emin=-9.0,
emax=16.0
/
We run:
dos.x < pp.dos.silicon.in > pp.dos.silicon.out
The DOS data in the si_dos.dat file that we specified in our input file. We
can plot the DOS:
import matplotlib.pyplot as plt
from matplotlib import rcParamsDefault
import numpy as np
%matplotlib inline
# load data
energy, dos, idos = np.loadtxt('../src/silicon/si_dos.dat', unpack=True)
# make plot
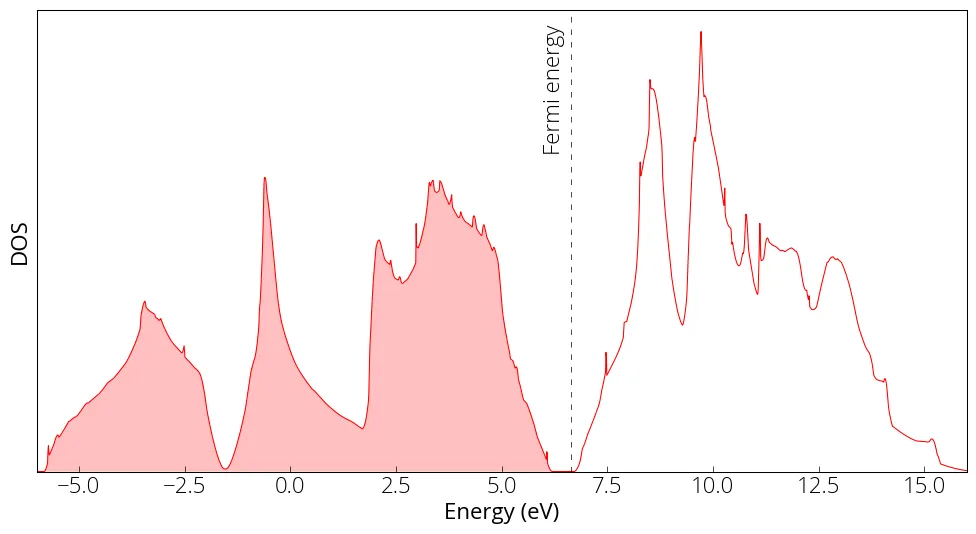
plt.figure(figsize = (12, 6))
plt.plot(energy, dos, linewidth=0.75, color='red')
plt.yticks([])
plt.xlabel('Energy (eV)')
plt.ylabel('DOS')
plt.axvline(x=6.642, linewidth=0.5, color='k', linestyle=(0, (8, 10)))
plt.xlim(-6, 16)
plt.ylim(0, )
plt.fill_between(energy, 0, dos, where=(energy < 6.642), facecolor='red', alpha=0.25)
plt.text(6, 1.7, 'Fermi energy', fontsize= med, rotation=90)
plt.show()
For a set of calculation, we must keep the prefix same. For example, the
nscf or bands calculation uses the wavefunction calculated by the
scf calculation. When performing different calculations, for example you
change a parameter and want to see the changes, you must use different output
folder or unique prefix for different calculations so that the outputs do not
get mixed.
Sometimes it is important to sample the point for DOS calculation (e.g., the conducting bands cross the Fermi surface only at point). In such cases, we need to use odd k-grid (e.g., 9✕9✕5).