DFT+U calculation
Electronic structure for transition metals (with localized or electrons) is not accurately described by standard DFT, and therefore the need for DFT+U formulation.
&SYSTEM
...
lda_plus_u = .TRUE.
Hubbard_u(i) = 2.0
...
/
Here i refers to the atomic index in the &ATOMIC_SPECIES card corresponding
to each ntyp. We can specify Hubbard_u(i) corresponding to more than one
atom in separate lines.
There is also implementation in QE. represents on-site exchange interaction. Number of terms depends on the manifold of localized electrons. For , we have 1; for , we have 2; and for , we have 3 terms.
...
lda_plus_u = .TRUE.
lda_plus_u_kind = 1
Hubbard_u(i) = U
Hubbard_J(k, i) = J_{ki}
...
If you add Hubbard_u for elements that is not implemented to have term
in QE, you might see a "pseudopotential not yet inserted" error.
Changes to input syntax in v7.1
Starting from Quantum Espresso version 7.1, there are changes to input syntax
for DFT+U calculations. In the new version, instead of defining the necessary
DFT+U parameters, now there is a new Hubbard card.
&system
...
- lda_plus_u = .true.,
- lda_plus_u_kind = 0,
- U_projection_type = 'atomic',
- Hubbard_U(1) = 4.6
- Hubbard_U(2) = 4.6
...
/
+ HUBBARD (ortho-atomic)
+ U Fe1-3d 4.6
+ U Fe2-3d 4.6
Please refer to the qe-x.x/Doc/Hubbard_input.pdf for details.
DFT calculation for FeO
We will first perform the standard DFT calculation.
- Perform the SCF calculation:
pw.x -in feo_scf.in > feo_scf.out
- Perform NSCF calculation with denser k-grid:
pw.x -in feo_nscf.in > feo_nscf.out
- Perform P-DOS calculation:
projwfc.x -in feo_projwfc.in > feo_projwfc.out
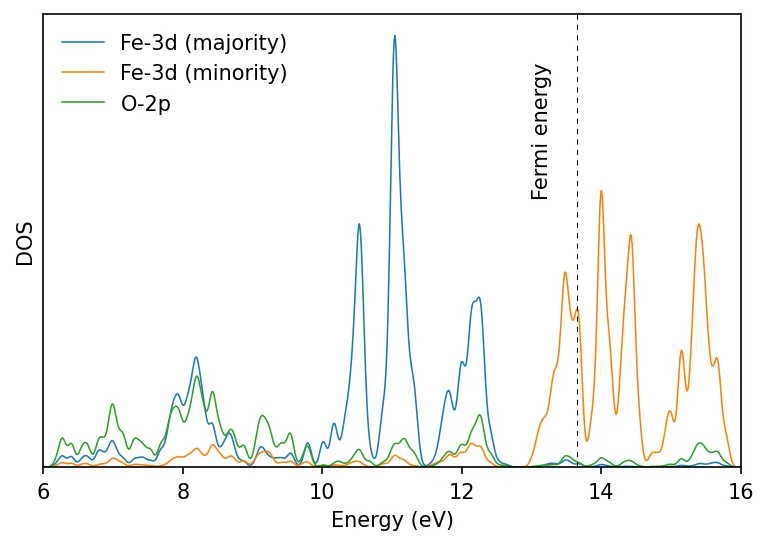
This gives us metallic density of states. In practice we get insulating FeO.
Calculating Hubbard U
&inputhp
prefix = 'FeO'
outdir = './tmp/'
nq1 = 1, nq2 = 1, nq3 = 1
/
Perform a linear-response calculation using hp.x program:
hp.x -in feo_hp.in > feo_hp.out
Check the file FeO.Hubbard_parameters.dat.
-
We need to check the convergence against q-mesh (as well as k-mesh in SCF calculation). Here mesh is used. Important:
lda_plus_umust be set to.true.during the SCF calculation, may be set to zero. -
We can update the obtained value in our SCF calculation, and repeat linear response calculation until we have reached self consistency in value.
-
To go even further one can check the convergence of geometry during updates.
-
There is also inter-site Hubbard correction DFT+U+V calculation. The results could be more closer to hybrid functionals like GW. The can also be calculated using Quantum Espresso hp.x code.
-
Obtained value of depends on pseudopotential, Hubbard manifold (whether atomic, ortho-atomic etc.).
Calculating the self-consistent Hubbard parameter involves an iterative
workflow that begins with a standard self-consistent field (SCF) calculation
using a tiny initial value . Next, Density Functional
Perturbation Theory (DFPT) is performed via the hp.x module to calculate a
new linear-response Hubbard parameter . This newly obtained
is then used to perform a structural optimization (relaxation) of the crystal
system. Finally, the calculated is compared against the initial
; if they have converged to a matching value, self-consistency is
successfully reached. If they do not match, the loop repeats by feeding the new
value and the updated CELL_PARAMETERS back into a fresh SCF
calculation until convergence is achieved. A convergence tolerance of 10
meV/site for is typically recommended.
The above hp.x code is not suitable for closed cell systems (e.g., fully occupied d-shell element), in such cases this linear response method gives unrealistically large value.
DFT+U calculation
We repeat the calculation after setting in the &SYSTEM card:
Hubbard_U(1) = 4.6
Hubbard_U(2) = 4.6
We repeat the above calculation and plot the results. Now we find insulating ground state.
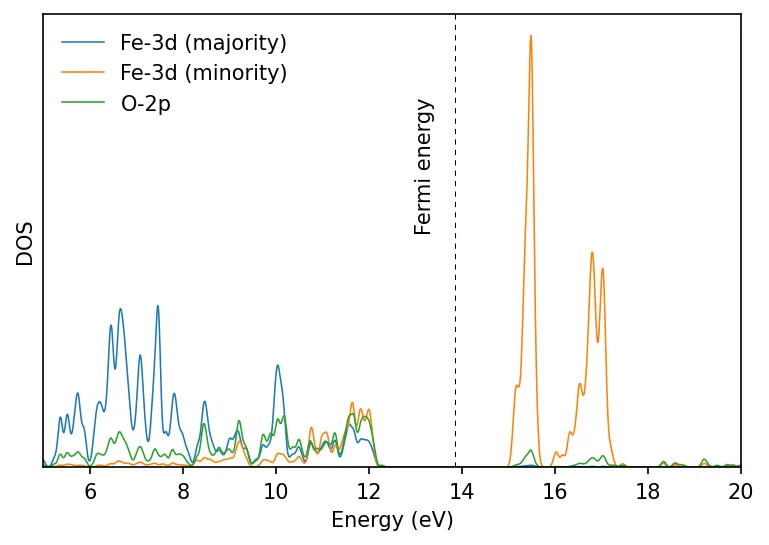
U_projection_type = 'ortho-atomic' might give more realistic result than the
default 'atomic'.
When performing calculation, the ground state might get stuck in a
local minimum, in such cases we need to provide starting_ns_eigenvalue to
help calculation reach desired/actual ground state. Please see these slides by
Dr. Iurii Timrov for a relevant example.
Here we have plotted the lpdos (local density of states). If we want to know
the contribution of ect., we can find them from
the pdos columns. Also there arise important Lowdin charges information in the
feo_projwfc.out file.