DOS and bandstructure for Aluminum (metal)
Variable cell relaxation
First we are going to relax the cell and choose appropriate lattice constant for our chosen pseudo potential. In case of metals, it is important to provide smearing parameters in the input file.
&CONTROL
calculation= 'vc-relax',
prefix= 'al',
outdir= '/tmp/'
pseudo_dir = '../pseudos/'
etot_conv_thr= 1e-6,
forc_conv_thr= 1e-5
/
&SYSTEM
ibrav= 2,
celldm(1)= 7.652,
nat= 1,
ntyp= 1,
ecutwfc = 50,
ecutrho= 500,
occupations= 'smearing',
smearing= 'gaussian',
degauss= 0.01
/
&ELECTRONS
conv_thr= 1e-8
/
&IONS
/
&CELL
cell_dofree= 'ibrav'
/
ATOMIC_SPECIES
Al 26.981539 Al.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (alat)
Al 0.00 0.00 0.00
K_POINTS (automatic)
10 10 10 0 0 0
We run pw.x to perform variable cell relaxation calculation:
pw.x < al_vc_relax.in > al_vc_relax.out
Now you may open the output file in vi editor and invoke search by pressing
/ and type Final enthalpy You will find the final lattice parameters below
it.
Self consistent field (SCF) calculation
We obtain relaxed lattice constant = 7.652 * 0.498611683 / 0.5 = 7.63075 Bohr. We will use this value for our next step, self consistent calculation.
&CONTROL
calculation= 'scf',
restart_mode= 'from_scratch',
prefix= 'al',
outdir= '/tmp/',
pseudo_dir= '../pseudos/'
/
&SYSTEM
ibrav= 2,
celldm(1) = 7.63075,
nat= 1,
ntyp= 1,
ecutwfc= 50,
ecutrho= 500,
occupations= 'smearing',
smearing= 'gaussian',
degauss= 0.01
/
&ELECTRONS
conv_thr= 1e-8
/
ATOMIC_SPECIES
Al 26.981539 Al.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (alat)
Al 0.00 0.00 0.00
K_POINTS (automatic)
10 10 10 0 0 0
We run our self consistent calculation:
pw.x < al_scf.in > al_scf.out
Non-self consistent field calculation
Inspect the output file, and let's proceed to next step non-self consistent calculation:
&CONTROL
calculation= 'nscf',
restart_mode= 'from_scratch',
prefix= 'al',
outdir= '/tmp/',
pseudo_dir= '../pseudos/'
/
&SYSTEM
ibrav= 2,
celldm(1) = 7.63075,
nat= 1,
ntyp= 1,
ecutwfc= 50,
ecutrho= 500,
occupations= 'smearing',
smearing= 'gaussian',
degauss= 0.01
/
&ELECTRONS
conv_thr= 1e-8
/
ATOMIC_SPECIES
Al 26.981539 Al.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (alat)
Al 0.00 0.00 0.00
K_POINTS (automatic)
40 40 40 0 0 0
Note the changes in input file. The calculation changed to nscf and we are now
using a higher number of k-points grid.
pw.x < al_nscf.in > al_nscf.out
Density of states
Next we go ahead with our density of states calculation:
&DOS
prefix= 'al',
outdir= '/tmp/',
fildos= 'al_dos.dat',
emin= -10,
emax= 35
/
We run dos.x with DOS inputs:
dos.x < al_dos.in > al_dos.out
Note from our al_nscf.out that our Fermi energy is at 7.9421 eV. We plot our
density of states:
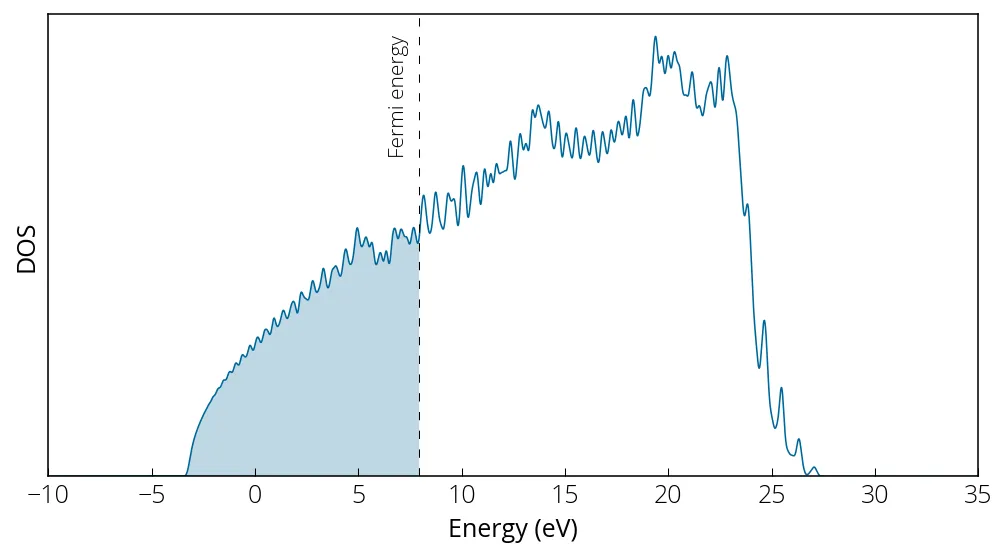
Bandstructure calculation
We prepare the input file the same as the case of our previous example silicon:
&CONTROL
calculation= 'bands',
restart_mode= 'from_scratch',
prefix= 'al',
outdir= '/tmp/',
pseudo_dir= '../pseudos/'
/
&SYSTEM
ibrav= 2,
celldm(1) = 7.63075,
nat= 1,
ntyp= 1,
ecutwfc= 50,
ecutrho= 500,
occupations= 'smearing',
smearing= 'gaussian',
degauss= 0.01
/
&ELECTRONS
conv_thr= 1e-8
/
ATOMIC_SPECIES
Al 26.981539 Al.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (alat)
Al 0.00 0.00 0.00
K_POINTS {crystal_b}
5
00.000 0.500 00.000 20 !L
00.000 0.000 00.000 30 !G
-0.500 0.000 -0.500 10 !X
-0.375 0.250 -0.375 30 !U
00.000 0.000 00.000 20 !G
Followed by run pw.x:
pw.x < al_bands.in > al_bands.out
Now we proceed with post-processing:
&BANDS
prefix = 'al'
outdir = '/tmp/'
filband = 'al_bands.dat'
/
And run bands.x:
bands.x < al_bands_pp.in > al_bands_pp.out
We obtain the following bandstructure:
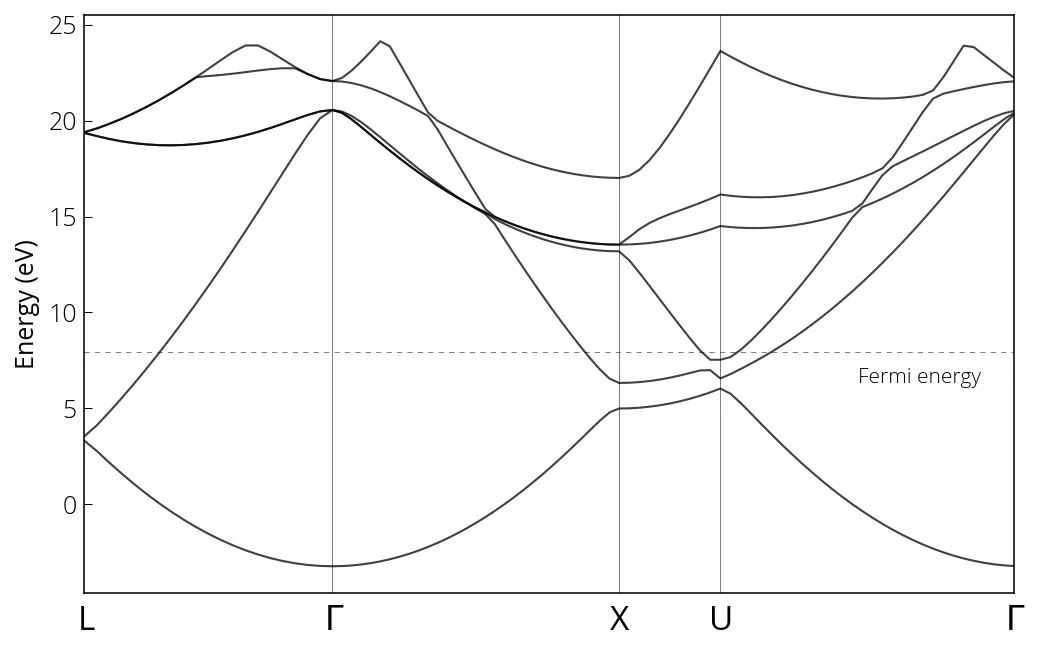
Importance of smearing in convergence
Smearing is a technique used for suppressing unstable electron density in the calculation of metals. Such a problem occurs in metals (and semimetals) because the valence bands that cross Fermi level are partially occupied. Due to numerical accuracy, the electrons may occupy the unoccupied states during some iterations, making the algorithm unstable. In order to stablize the algorithm without using excessive number of k-points, smearing technique is used, which replaces the occupation number (either 0 or 1) is replaced by a smoothly varying function of energy. Such a smearing function could be Fermi Dirac distribution, instead of a step function (T = 0 K), we can use the finite temperature form.
Below we will test the convergence using PWTK against the number of k-points, three different smearing functions (Gauss, Methfessel-Paxton, and Marzari-Vanderbilt), and for various smearing values.
pwtk al.degauss.pwtk
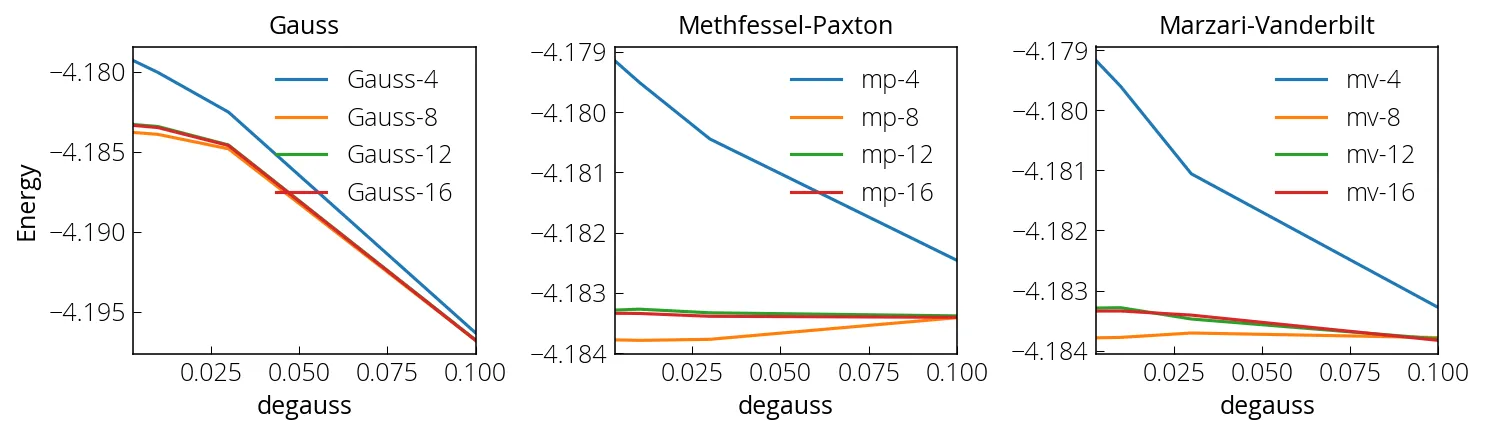
We see that the m-v and m-p broadening allow for faster and smother
convergence while depending less on degauss value than Gaussian broadening.
The number suffix next to the legend labels are number of uniform k-points in
Monkhorst-Plank grid.