Bandstructure of GaAs
Now that we have calculated the bandstructure of silicon (semiconductor) and aluminum (metal), let us proceed with a compound which has two different atoms. We follow the steps like before:
First check the lattice constant with cell relaxation according to our chosen pseudo potential. We use that lattice constant in our next steps. Our lattice constant = 10.6867 * 0.508176602 / 0.5 = 10.861462.
pw.x < pw.relax.GaAs.in > pw.relax.GaAs.out
Perform self consistent field calculation:
pw.x < pw.scf.GaAs.in > pw.scf.GaAs.out
Give denser k-points and perform non-self consistent calculation. This step is only necessary if you need to obtain density of states.
pw.x < pw.nscf.GaAs.in > pw.nscf.GaAs.out
Perform bands calculation
pw.x < pw.bands.GaAs.in > pw.bands.GaAs.out
Post process the data and plot the bandstructure.
bands.x < pp.bands.GaAs.in > pp.bands.GaAs.out
If everything goes well, you will get the bandstructure as below:
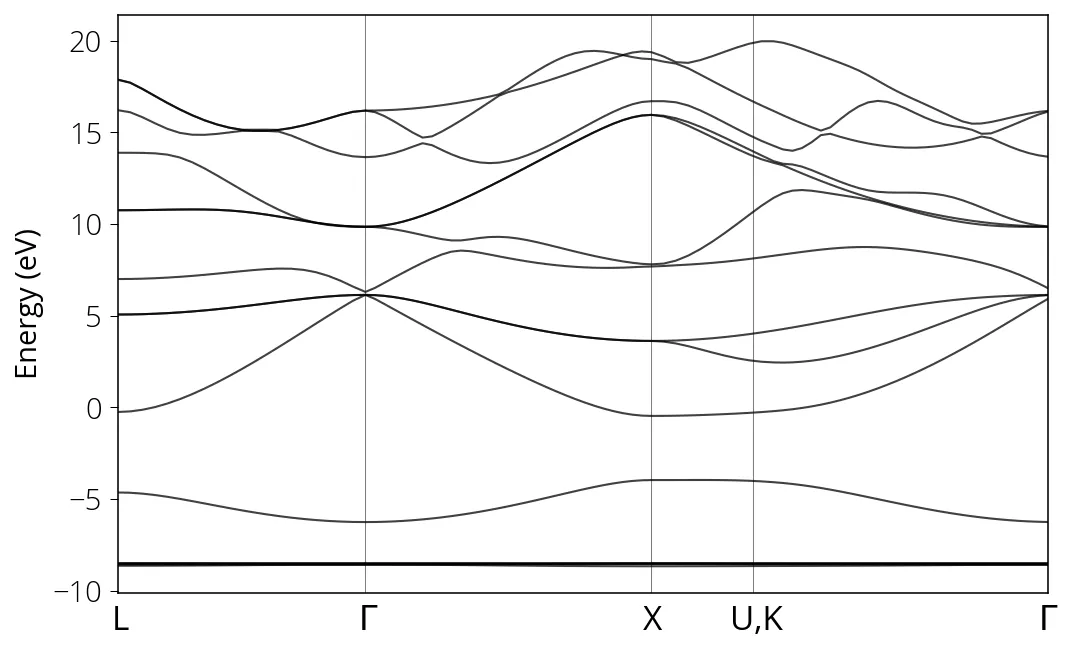
Sometimes a calculation with the same inputs converges in one computer, while fails in another due to library configuration or even due to floating point approximations. The final output numbers will always vary slightly for different machines, or even among different runs in the same machine. Also check the Quantum Espresso version for reproducibility.
Also see the bandstructure of GaAs with SOC.